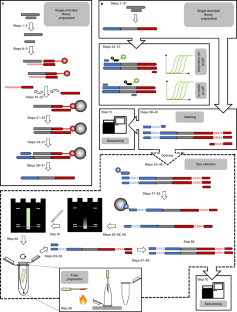
Manual and automated preparation of single-stranded DNA libraries for the sequencing of DNA from ancient biological remains and other sources of highly degraded DNA
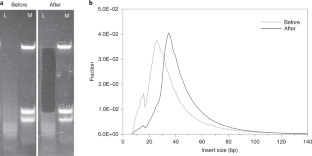
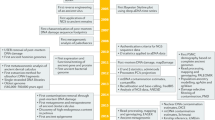
It has been shown that highly fragmented DNA is most efficiently converted into DNA libraries for sequencing if both strands of the DNA fragments are processed independently. We present an updated protocol for library preparation from single-stranded DNA, which is based on the splinted ligation of an adapter oligonucleotide to the 3′ ends of single DNA strands, the synthesis of a complementary strand using a DNA polymerase and the addition of a 5′ adapter via blunt-end ligation. The efficiency of library preparation is determined individually for each sample using a spike-in oligonucleotide. The whole workflow, including library preparation, quantification and amplification, requires two work days for up to 16 libraries. Alternatively, we provide documentation and electronic protocols enabling automated library preparation of 96 samples in parallel on a Bravo NGS Workstation (Agilent Technologies). After library preparation, molecules with uninformative short inserts (shorter than ~30−35 base pairs) can be removed by polyacrylamide gel electrophoresis if desired.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
265,23 € per year
only 22,10 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

A highly efficient scheme for library preparation from single-stranded DNA
Article Open access 25 August 2023

Maximizing efficiency in sedimentary ancient DNA analysis: a novel extract pooling approach
Article Open access 20 August 2024
Ancient DNA analysis
Article 11 February 2021
Data availability
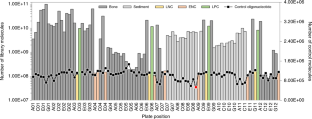
All raw data points underlying Fig. 3 are listed in Supplementary Table 1.
Code availability
Electronic protocol files for automated library preparation and auxiliary files are available at the Zenodo website (https://zenodo.org/record/3631147).
References
- Meyer, M. et al. A high-coverage genome sequence from an archaic Denisovan individual. Science338, 222–226 (2012). CASPubMedPubMed CentralGoogle Scholar
- Gansauge, M. T. & Meyer, M. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat. Protoc.8, 737–748 (2013). PubMedGoogle Scholar
- Prüfer, K. et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature505, 43–49 (2014). PubMedGoogle Scholar
- Prüfer, K. et al. A high-coverage Neandertal genome from Vindija Cave in Croatia. Science358, 655–658 (2017). PubMedPubMed CentralGoogle Scholar
- Mafessoni, F. et al. A high-coverage Neandertal genome from Chagyrskaya Cave. Preprint at https://www.biorxiv.org/content/10.1101/2020.03.12.988956v1 (2020).
- Fu, Q. et al. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature514, 445–449 (2014). CASPubMedPubMed CentralGoogle Scholar
- Dabney, J. et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl Acad. Sci. USA110, 15758–15763 (2013). CASPubMedPubMed CentralGoogle Scholar
- Meyer, M. et al. A mitochondrial genome sequence of a hominin from Sima de los Huesos. Nature505, 403–406 (2014). CASPubMedGoogle Scholar
- Meyer, M. et al. Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins. Nature531, 504–507 (2016). CASPubMedGoogle Scholar
- Slon, V. et al. Neandertal and Denisovan DNA from Pleistocene sediments. Science356, 605–608 (2017). CASPubMedGoogle Scholar
- Korlević, P. et al. Reducing microbial and human contamination in DNA extractions from ancient bones and teeth. Biotechniques59, 87–93 (2015). PubMedGoogle Scholar
- Gansauge, M. T. et al. Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Res.45, e79 (2017). CASPubMedPubMed CentralGoogle Scholar
- Gansauge, M. T. & Meyer, M. A method for single-stranded ancient DNA library preparation. Methods Mol. Biol. 1963, 75–83 (2019).
- Glocke, I. & Meyer, M. Extending the spectrum of DNA sequences retrieved from ancient bones and teeth. Genome Res.27, 1230–1237 (2017). CASPubMedPubMed CentralGoogle Scholar
- de Filippo, C., Meyer, M. & Prüfer, K. Quantifying and reducing spurious alignments for the analysis of ultra-short ancient DNA sequences. BMC Biol.16, 121 (2018). PubMedPubMed CentralGoogle Scholar
- Bennett, E. A. et al. Library construction for ancient genomics: single strand or double strand? Biotechniques56, 289–290 (2014). CASPubMedGoogle Scholar
- Wales, N. et al. New insights on single-stranded versus double-stranded DNA library preparation for ancient DNA. Biotechniques59, 368–371 (2015). CASPubMedGoogle Scholar
- Meyer, M. & Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, pdb prot5448 (2010).
- Caroe, C. et al. Single-tube library preparation for degraded DNA. Methods Ecol. Evol.9, 410–419 (2018). Google Scholar
- Briggs, A. W. et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl Acad. Sci. USA104, 14616–14621 (2007). CASPubMedPubMed CentralGoogle Scholar
- Skoglund, P. et al. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc. Natl Acad. Sci. USA111, 2229–2234 (2014). CASPubMedPubMed CentralGoogle Scholar
- Gansauge, M. T. & Meyer, M. Selective enrichment of damaged DNA molecules for ancient genome sequencing. Genome Res.24, 1543–1549 (2014). CASPubMedPubMed CentralGoogle Scholar
- Bokelmann, L. et al. A genetic analysis of the Gibraltar Neanderthals. Proc. Natl Acad. Sci. USA116, 15610–15615 (2019). CASPubMedPubMed CentralGoogle Scholar
- Stiller, M. et al. Single-strand DNA library preparation improves sequencing of formalin-fixed and paraffin-embedded (FFPE) cancer DNA. Oncotarget7, 59115–59128 (2016). PubMedPubMed CentralGoogle Scholar
- Snyder, M. W., Kircher, M., Hill, A. J., Daza, R. M. & Shendure, J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell164, 57–68 (2016). CASPubMedPubMed CentralGoogle Scholar
- Burnham, P. et al. Single-stranded DNA library preparation uncovers the origin and diversity of ultrashort cell-free DNA in plasma. Sci. Rep. 6, 27859 (2016).
- Turchinovich, A. et al. Capture and Amplification by Tailing and Switching (CATS). An ultrasensitive ligation-independent method for generation of DNA libraries for deep sequencing from picogram amounts of DNA and RNA. RNA Biol.11, 817–828 (2014). CASPubMedPubMed CentralGoogle Scholar
- Karlsson, K. et al. Amplification-free sequencing of cell-free DNA for prenatal non-invasive diagnosis of chromosomal aberrations. Genomics105, 150–158 (2015). CASPubMedGoogle Scholar
- Raine, A., Manlig, E., Wahlberg, P., Syvanen, A. C. & Nordlund, J. SPlinted Ligation Adapter Tagging (SPLAT), a novel library preparation method for whole genome bisulphite sequencing. Nucleic Acids Res.45, e36 (2017). PubMedGoogle Scholar
- Wu, J., Dai, W., Wu, L. & Wang, J. SALP, a new single-stranded DNA library preparation method especially useful for the high-throughput characterization of chromatin openness states. BMC Genomics19, 143 (2018). PubMedPubMed CentralGoogle Scholar
- Ding, J., Taylor, M. S., Jackson, A. P. & Reijns, M. A. Genome-wide mapping of embedded ribonucleotides and other noncanonical nucleotides using emRiboSeq and EndoSeq. Nat. Protoc.10, 1433–1444 (2015). CASPubMedPubMed CentralGoogle Scholar
- Petryk, N. et al. Replication landscape of the human genome. Nat. Commun.7, 10208 (2016). CASPubMedPubMed CentralGoogle Scholar
- Tin, M. M., Economo, E. P. & Mikheyev, A. S. Sequencing degraded DNA from non-destructively sampled museum specimens for RAD-tagging and low-coverage shotgun phylogenetics. PLoS ONE9, e96793 (2014). PubMedPubMed CentralGoogle Scholar
- Troll, C. J. et al. A ligation-based single-stranded library preparation method to analyze cell-free DNA and synthetic oligos. BMC Genomics20, 1023 (2019). CASPubMedPubMed CentralGoogle Scholar
- Knapp, M., Clarke, A. C., Horsburgh, K. A. & Matisoo-Smith, E. A. Setting the stage - building and working in an ancient DNA laboratory. Ann. Anat.194, 3–6 (2012). CASPubMedGoogle Scholar
- Kampmann, M. L., Borsting, C. & Morling, N. Decrease DNA contamination in the laboratories. Forensic Sci. Int. Genet.6, E577–E578 (2017). Google Scholar
- Rohland, N., Glocke, I., Aximu-Petri, A. & Meyer, M. Extraction of highly degraded DNA from ancient bones, teeth and sediments for high-throughput sequencing. Nat. Protoc.13, 2447–2461 (2018). CASPubMedGoogle Scholar
- Prüfer, K. snpAD: an ancient DNA genotype caller. Bioinformatics34, 4165–4171 (2018). PubMedPubMed CentralGoogle Scholar
- Varshney, U. & van de Sande, J. H. Specificities and kinetics of uracil excision from uracil-containing DNA oligomers by Escherichia coli uracil DNA glycosylase. Biochemistry30, 4055–4061 (1991). CASPubMedGoogle Scholar
- Burbano, H. A. et al. Targeted investigation of the Neandertal genome by array-based sequence capture. Science328, 723–725 (2010). CASPubMedPubMed CentralGoogle Scholar
- Fu, Q. et al. DNA analysis of an early modern human from Tianyuan Cave, China. Proc. Natl Acad. Sci. USA110, 2223–2227 (2013). CASPubMedPubMed CentralGoogle Scholar
- Haak, W. et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature522, 207–211 (2015). CASPubMedPubMed CentralGoogle Scholar
- Kircher, M., Sawyer, S. & Meyer, M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res40, e3 (2012). CASPubMedGoogle Scholar
- Dabney, J. & Meyer, M. Length and GC-biases during sequencing library amplification: a comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. Biotechniques52, 87–94 (2012). CASPubMedGoogle Scholar
- Thompson, J. R., Marcelino, L. A. & Polz, M. F. Heteroduplexes in mixed-template amplifications: formation, consequence and elimination by ‘reconditioning PCR’. Nucleic Acids Res.30, 2083–2088 (2002). CASPubMedPubMed CentralGoogle Scholar
- Renaud, G., Stenzel, U. & Kelso, J. leeHom: adaptor trimming and merging for Illumina sequencing reads. Nucleic Acids Res.42, e141 (2014). PubMedPubMed CentralGoogle Scholar
- Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics25, 1754–1760 (2009). CASPubMedPubMed CentralGoogle Scholar
Acknowledgements
We thank L. Bokelmann, E. Essel, L. Lippik, J. Richter, B. Schellbach and A. Weihmann for help in the lab, D. Massilani, S. Pääbo, B. Vernot and E. Zavala for helpful discussions, I. Bünger for help with installing software, J. Kelso and J. Visagie for help with data processing and L. Jauregui for comments on the manuscript. This work was funded by the Max Planck Society.
Author information
Authors and Affiliations
- Department of Evolutionary Genetics, Max Planck Institute for Evolutionary Anthropology, Leipzig, Germany Marie-Theres Gansauge, Ayinuer Aximu-Petri, Sarah Nagel & Matthias Meyer
- Marie-Theres Gansauge
